Maetrics has launched a new white paper highlighting the critical importance for Class I medical device manufacturers to comply with the EU Medical Device Regulation (MDR) coming into force next year.
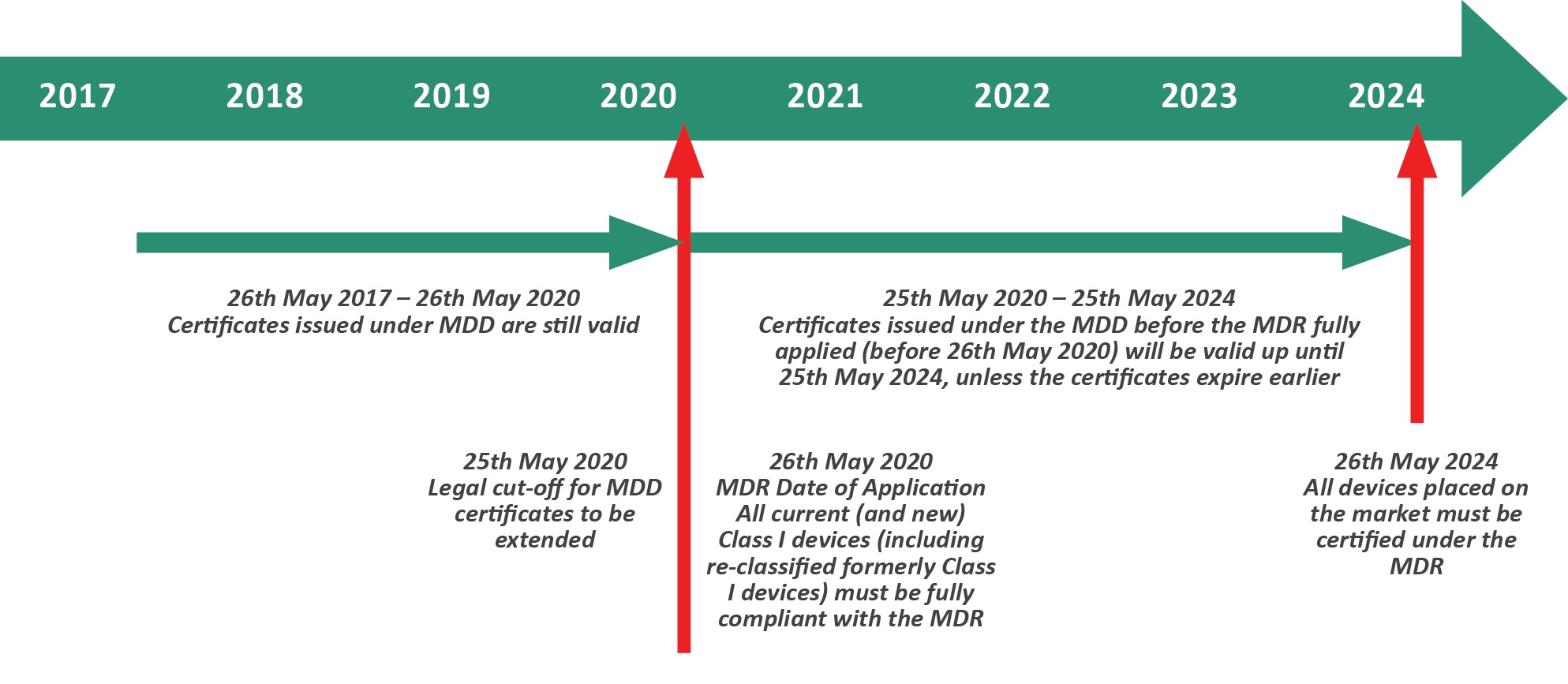
The MDR transition period ends on 26 May next year, at which point any non-compliant manufacturers will be restricted from selling and distributing their healthcare products on the EU market.
Our aim is to ensure that Class I medical device manufacturers are fully aware of what is at stake if they don’t comply with the MDR as soon as possible
This new white paper has been created to serve as a valuable tool for device manufacturers to help ensure they maintain EU market access.
Its aim is to raise awareness of the pressing issue of dealing with reclassification and achieving timely compliance under the MDR, providing guidance and practical steps that Class I manufacturers should take as soon as possible.
Key points include:
- The MDR contains new stringent rules on classification, causing many current Class I devices to be up-classified
- More Class I products will require Notified Body review to comply with the regulation and have the appropriate certificates for CE Marking
- Only devices which were CE marked under the authority of a Notified Body through the MDD will have a grace period until up to May 2024 to continue selling their devices in Europe
- Any Class I device which was previously self certified and now needs Notified Body review must have a new MDR-compliant CE Mark by 26 May 2020
Any manufacturer unaware their products are being up-classified risks not having the appropriate post-market and clinical data to submit to their Notified Body in order to ensure certification under the MDR by the deadline.
And a delay in tackling MDR conformance could result in further delays due to increasing Notified Body capacity issues, as the commission scrambles to redesignate the Notified Bodies under the MDR.
Peter Rose, managing director at Maetrics, said: “Our aim is to ensure that Class I medical device manufacturers are fully aware of what is at stake if they don’t comply with the MDR as soon as possible.
“It has long been established that the MDR does not currently allow for any grandfathering of products, meaning all products must be fully compliant to the MDR as if they were a new product.
It has long been established that the MDR does not currently allow for any grandfathering of products, meaning all products must be fully compliant to the MDR as if they were a new product
“Previous good historical performance, even if a product has been on the EU market for 20-plus years without a complaint, is no guarantee of compliance for these manufacturers.
“Manufacturers need to be verifying the class which their devices fall into with the utmost urgency due to the sheer quantity of devices which have been up-classified under the MDR.
“Taking a pro-active approach to compliance is essential to be ready for EU MDR come May 2020.”
He added: “Even if a Class I medical device has not been up-classified, it must still be fully compliant to the MDR by the May 2020 deadline. This includes having a Quality Management System (QMS) in accordance with Article 10, paragraph 9. This effectively means that Class I manufacturers should implement an EN ISO 13485:2016 QMS. Our clients’ experience shows that Certification Bodies are asking to see Clinical Evaluations as a part of the QMS audit.”





